Category: Manual
7 附录
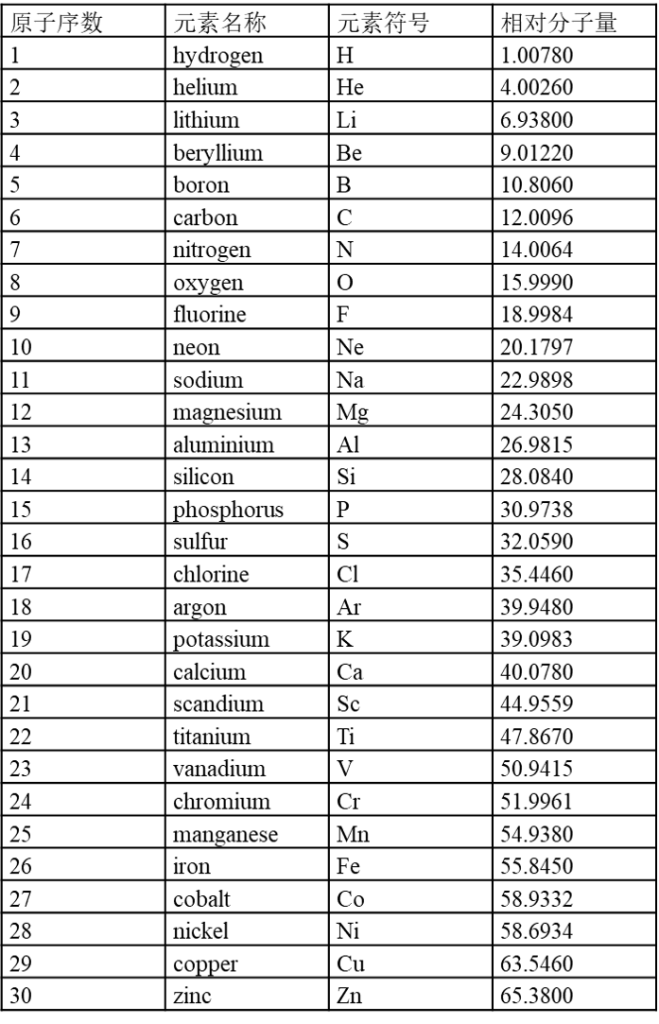
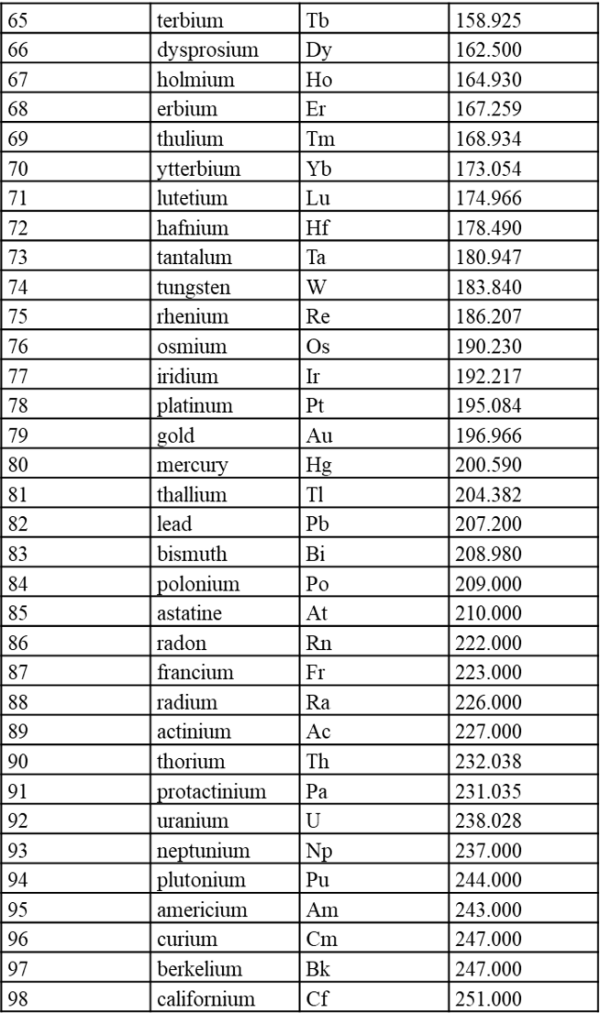
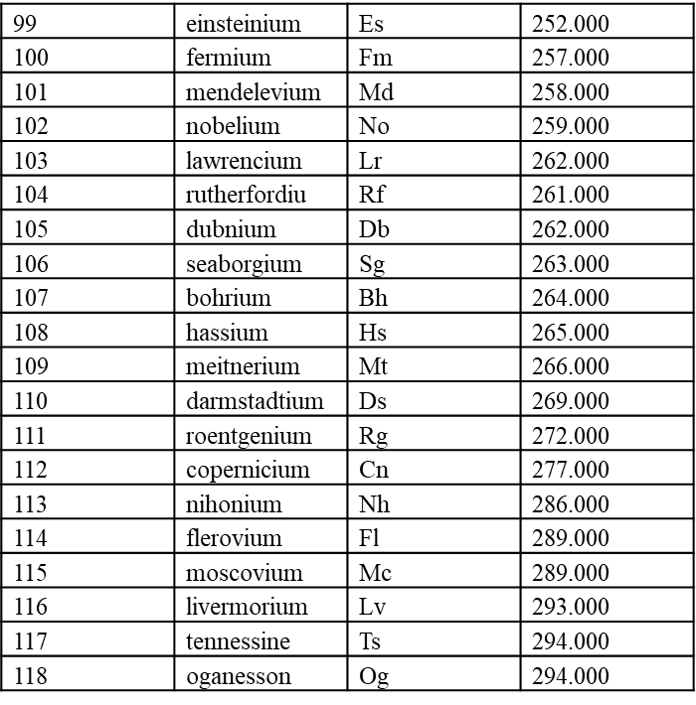
7.1 内置元素周期表参数
7.2 内置常见分子热力学参数
% Define the default thermodynamics data in the file Thermodynamics.data
% ‘%’ and ‘!’ for comments
% Key words: CF, TR, AH, FQ, EF
% CF: the chemical formula, TR: the temperature range, AH: the shomate parameters [A – H]
% FQ: the frequency, EF: the experimental formation energy
CF = H2; % 1;
EF = [ -0.272839]; FQ = [ 4401.00];
TR = [ 298.00, 1000.00]; AH = [ 33.066178, -11.363417, 11.432816, -2.772874, -0.158558, -9.980797, 172.707974, 0.000000];
TR = [ 1000.00, 2500.00]; AH = [ 18.563083, 12.257357, -2.859786, 0.268238, 1.977990, -1.147438, 156.288133, 0.000000];
TR = [ 2500.00, 6000.00]; AH = [ 43.413560, -4.293079, 1.272428, -0.096876, -20.533862, -38.515158, 162.081354, 0.000000];
CF = CH4; % 2;
EF = [ -1.865940]; FQ = [ 2917.00, 1534.00, 1534.00, 3019.00, 3019.00, 3019.00, 1306.00, 1306.00, 1306.00];
TR = [ 298.00, 1300.00]; AH = [ -0.703029, 108.477300, -42.521570, 5.862788, 0.678565, -76.843760, 158.716300, -74.873100];
TR = [ 1300.00, 1600.00]; AH = [ 85.812170, 11.264670, -2.114146, 0.138190, -26.422210, -153.532700, 224.414300, -74.873100];
CF = CO; % 3;
EF = [ -1.314068]; FQ = [ 2170.00];
TR = [ 298.00, 1300.00]; AH = [ 25.567590, 6.096130, 4.054656, -2.671201, 0.131021, -118.008900, 227.366500, -110.527100];
TR = [ 1300.00, 1600.00]; AH = [ 35.150700, 1.300095, -0.205921, 0.013550, -3.282780, -127.837500, 231.712000, -110.527100];
CF = H2O; % 4;
EF = [ -3.034448]; FQ = [ 3657.00, 1595.00, 3756.00];
TR = [ 298.00, 500.00]; AH = [ 36.303952, -24.112320, 63.641110, -38.952400, -0.013850, -252.066060, 237.394310, -241.826400];
TR = [ 500.00, 1700.00]; AH = [ 30.092000, 6.832514, 6.793435, -2.534480, 0.082139, -250.881000, 223.396700, -241.826400];
TR = [ 1700.00, 6000.00]; AH = [ 41.964260, 8.622053, -1.499780, 0.098119, -11.157640, -272.179700, 219.780900, -241.826400];
CF = CO2; % 5;
EF = [ -4.385561]; FQ = [ 1333.00, 2349.00, 667.00, 667.00];
TR = [ 298.00, 1200.00]; AH = [ 24.997350, 55.186960, -33.691370, 7.948387, -0.136638, -403.607500, 228.243100, -393.522400];
TR = [ 1200.00, 6000.00]; AH = [ 58.166390, 2.720075, -0.492289, 0.038844, -6.447293, -425.918600, 263.612500, -393.522400];
CF = O2; % 6;
EF = [ -0.097960]; FQ = [ 1580.00];
TR = [ 100.00, 700.00]; AH = [ 31.322340, -20.235310, 57.866400, -36.506240, -0.007374, -8.903471, 246.794500, 0.000000];
TR = [ 700.00, 2000.00]; AH = [ 30.032350, 8.772972, -3.988133, 0.788313, -0.741599, -11.324680, 236.166300, 0.000000];
TR = [ 2000.00, 6000.00]; AH = [ 20.911110, 10.720710, -2.020498, 0.146449, 9.245722, 5.337651, 237.618500, 0.000000];
CF = NH3; % 7;
EF = [ -1.298171]; FQ = [ 3337.00, 950.00, 3444.00, 3444.00, 1627.00, 1627.00];
TR = [ 298.00, 1400.00]; AH = [ 19.995630, 49.771190, -15.375990, 1.921168, 0.189174, -53.306670, 203.859100, -45.898060];
CF = N2; % 8;
EF = [ -0.146214]; FQ = [ 2359.00];
TR = [ 100.00, 500.00]; AH = [ 28.986410, 1.853978, -9.647459, 16.635370, 0.000117, -8.671914, 226.416800, 0.000000];
TR = [ 500.00, 2000.00]; AH = [ 19.505830, 19.887050, -8.598535, 1.369784, 0.527601, -4.935202, 212.390000, 0.000000];
CF = N2O; % 9;
EF = [ 0.590717]; FQ = [ 2224.00, 1285.00, 589.00];
TR = [ 298.00, 1400.00]; AH = [ 27.679880, 51.148980, -30.644540, 6.847911, -0.157906, 71.249340, 238.616400, 82.048240];
CF = NO2; % 10;
EF = [ 0.152695]; FQ = [ 1318.00, 1618.00, 750.00];
TR = [ 298.00, 1200.00]; AH = [ 16.108570, 75.895250, -54.387400, 14.307770, 0.239423, 26.174640, 240.538600, 33.095020];
CF = NO; % 11;
EF = [ 0.820336]; FQ = [ 1904.00];
TR = [ 298.00, 1200.00]; AH = [ 23.834910, 12.588780, -1.139011, -1.497459, 0.214194, 83.357830, 237.121900, 90.291140];
CF = NO3; % 12;
EF = []; FQ = [ 1450.00];
TR = [ 298.00, 1100.00]; AH = [ 11.223160, 166.388900, -148.445800, 47.405980, -0.176791, 61.008580, 221.767900, 71.128000];
TR = [ 1100.00, 6000.00]; AH = [ 82.274180, 0.487106, -0.098769, 0.006853, -6.264954, 29.223110, 326.252800, 71.128000];
CF = HNO2; % 13;
EF = []; FQ = [];
TR = [ 298.00, 1200.00]; AH = [ 24.899740, 91.375630, -64.846140, 17.920070, -0.134737, -88.135960, 254.267100, -76.734980];
CF = HNO3; % 14;
EF = []; FQ = [];
TR = [ 298.00, 1200.00]; AH = [ 19.632290, 153.959900, -115.837800, 32.879550, -0.249114, -146.881800, 247.704900, -134.306000];
CF = HCN; % 15;
EF = [ 0.948927]; FQ = [ 3311.00, 712.00, 712.00, 2097.00];
TR = [ 298.00, 1200.00]; AH = [ 32.693730, 22.592050, -4.369142, -0.407697, -0.282399, 123.481100, 233.259700, 135.143200];
CF = C2H4; % 16;
EF = [ -0.705013]; FQ = [ 3026.00, 1623.00, 1342.00, 1023.00, 3103.00, 1236.00, 949.00, 943.00, 3106.00, 826.00, 2989.00, 1444.00];
TR = [ 298.00, 1200.00]; AH = [ -6.387880, 184.401900, -112.971800, 28.495930, 0.315540, 48.173320, 163.156800, 52.466940];
CF = CH2O; % 17;
EF = [ -1.786501]; FQ = [ 2783.00, 1746.00, 1500.00, 2843.00, 1249.00, 1167.00];
TR = [ 298.00, 1500.00]; AH = [ 5.193767, 93.232490, -44.854570, 7.882279, 0.551175, -119.359100, 202.466300, -115.897200];
CF = CH3OH; % 18;
EF = [ -3.315495]; FQ = [ 3681.00, 3000.00, 2844.00, 1477.00, 1455.00, 1345.00, 1060.00, 1033.00, 2960.00, 1477.00, 1165.00, 200.00];
TR = [ 298.00, 1500.00]; AH = [ -1.084581, 153.246357, -79.530504, 16.471302, 0.522033, -205.897417, 199.189375, -201.000000];
CF = CH3CH2OH; % 19;
EF = [ -4.325727]; FQ = [ 3653.00, 2984.00, 2939.00, 2900.00, 1490.00, 1464.00, 1412.00, 1371.00, 1256.00, 1091.00, 1028.00, 888.00, 417.00, 2991.00, 2910.00, 1446.00, 1275.00, 1161.00, 812.00];
TR = [ 298.00, 1500.00]; AH = [ -4.736788, 271.961816, -169.349465, 43.738602, 0.246434, -244.628281, 203.332562, -234.800000];
CF = CH3CHO; % 20;
EF = [ -3.115742]; FQ = [ 3005.00, 2917.00, 2822.00, 1743.00, 1441.00, 1400.00, 1352.00, 1113.00, 919.00, 509.00, 2967.00, 1420.00, 867.00, 763.00, 150.00];
TR = [ 298.00, 1500.00]; AH = [ 4.803739, 185.920024, -99.108461, 20.614739, 0.277080, -174.756600, 220.002447, -166.190000];
CF = HCOOH; % 21;
EF = [ -4.736916]; FQ = [ 3570.00, 2943.00, 1770.00, 1387.00, 1229.00, 1105.00, 625.00, 1033.00, 638.00];
TR = [ 298.00, 1500.00]; AH = [ 3.802752, 153.662179, -84.640468, 16.297378, 0.277206, -385.165270, 212.969897, -379.000000];
CF = H2O2; % 22;
EF = [ -2.034197]; FQ = [ 3617.95, 1393.50, 877.93, 370.89, 3560.00, 1273.68];
TR = [ 298.00, 1500.00]; AH = [ 34.256670, 555.184450, -35.154430, 9.087440, -0.422157, -149.909800, -257.060600, -136.106400];
6 致谢
感谢国家自然科学基金委(21703067, 21622305, 21873028),国家重大研发项目(2018YFA0208602),中国博士后科学基金(2017M611471)对本项目的支持。
5 模拟示例
在这里,我们将以合成氨反应为动力学模型,给出3种代表性的例子,包括单条件、一维和二维条件的模拟情况,其对应的输入文件分别如下(INPUT_1_single、INPUT_2_curve、INPUT_3_map),可以看到,我们可以以非常简单明了的输入参数,实现各种复杂催化反应微动力学模拟,结果自动分析及可视化,包括速率、覆盖度、可逆度、敏感度、反应级数、主路径分析及其反应势能图和流程图等。
5.1 输入及输出文件概览
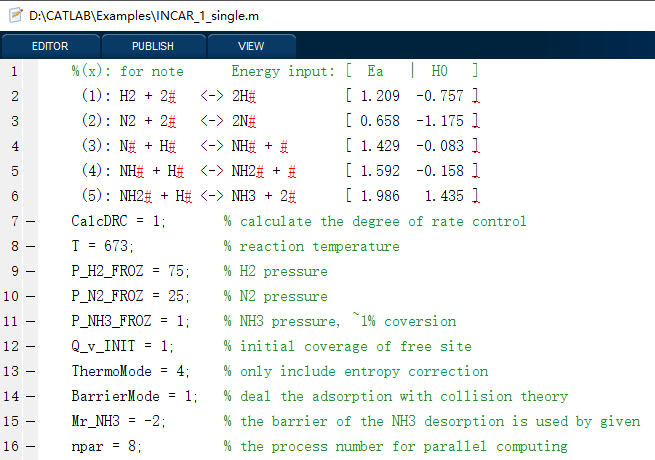
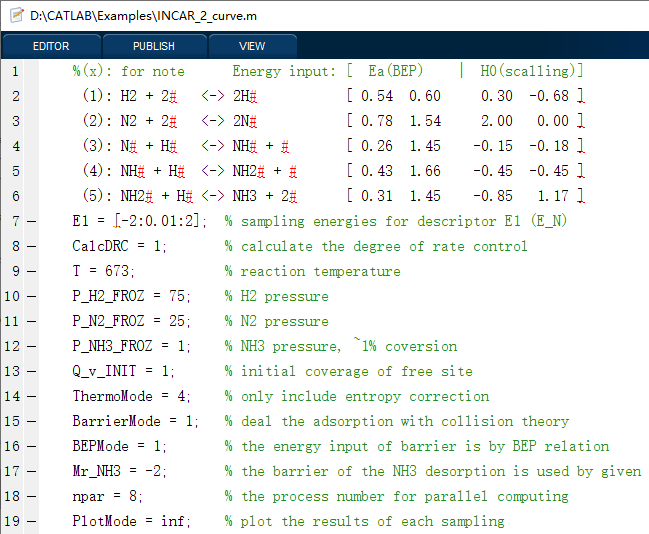
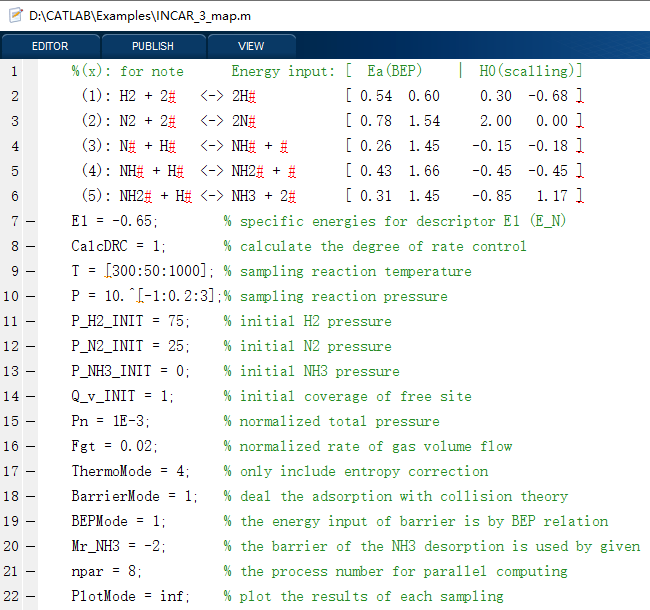
软件运行的截图如下:
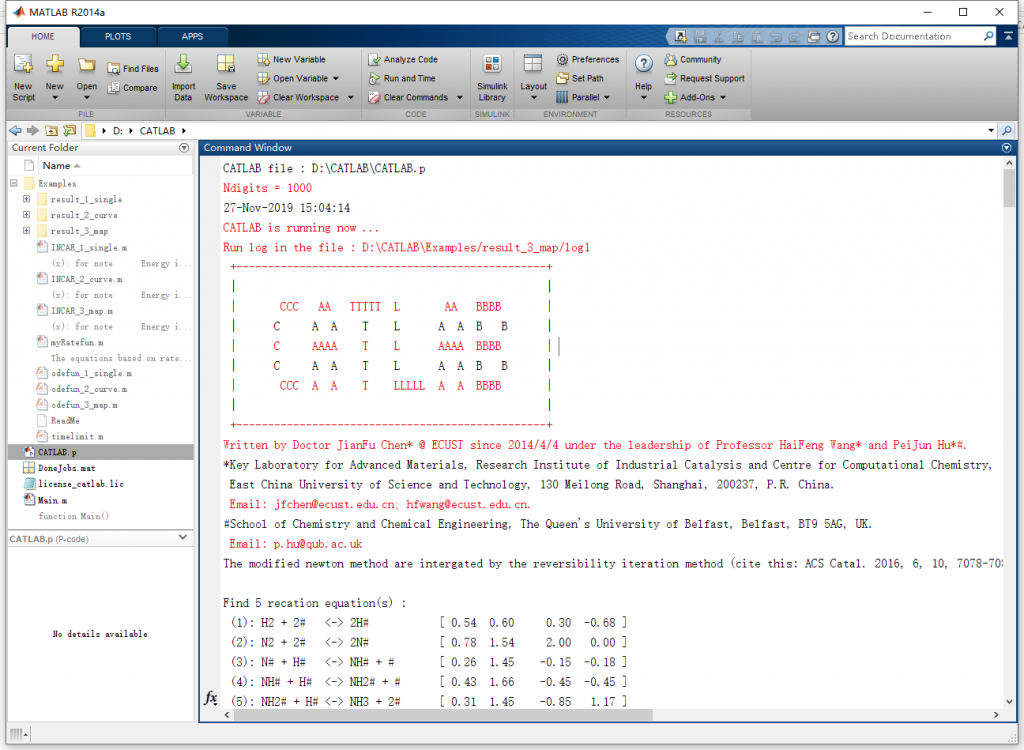
运行结束后,其对应的输出结果文件概览分别如下:
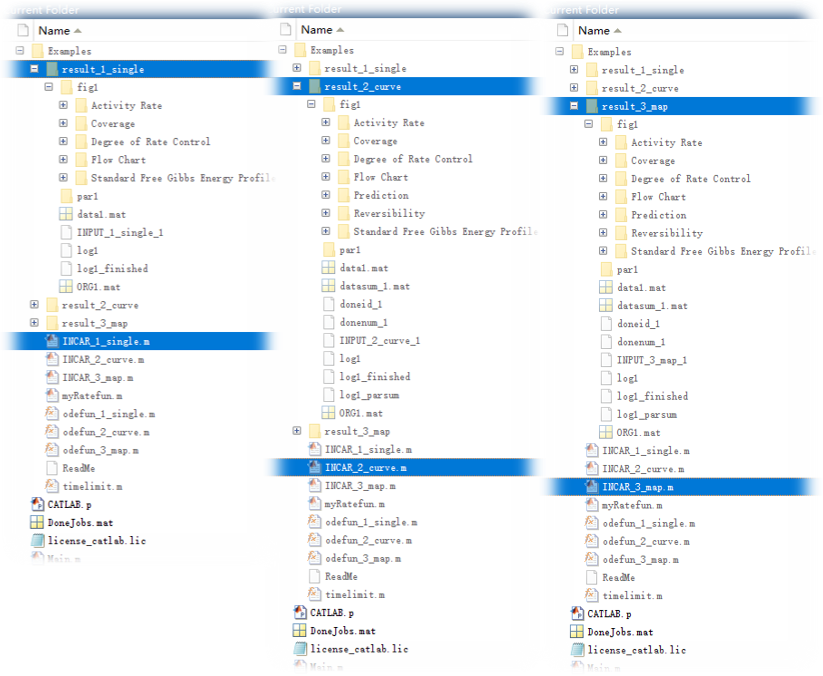
5.2 输出文件内容概览
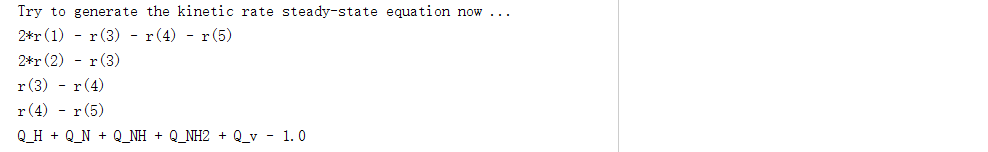
下面主要以单条件计算结果的输出文件log1为例,文件中包含自动生成的覆盖度、可逆度、基元反应速率、稳态方程组表达式:



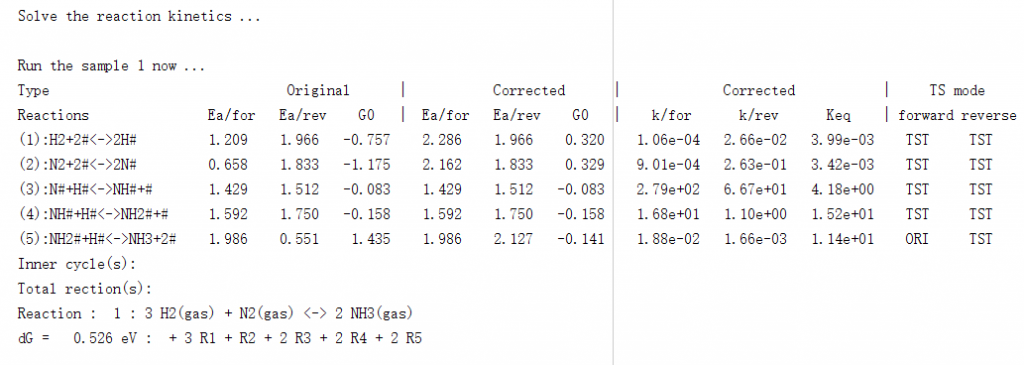
程序可以根据设置的参数,自动进行相应的热力学矫正和能垒处理,输出文件中包含了具体处理的方式、处理前后的数据等,供用户查看、使用或校验等:

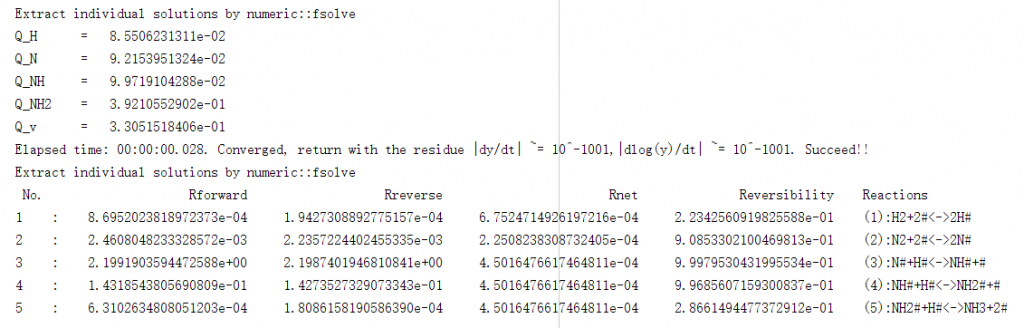
根据基元反应、反应能量和反应条件所对应的稳定方程,采用时间积分法、机器学习、粒子群优化算法等自动结合高精度的基于可逆度迭代的改进牛顿法,求解反应达到稳态时的覆盖度及其对应的速率:
进一步可以求解反应能垒、中间体敏感度,并自动分析和提取反应决速步骤、关键中间体:

还可以求解反应的级数,并自动分析和提取主要反应物种:

在程序运行的过程中,程序将会实时更新运行的模块(包括生成、求解、分析和绘图等)、进度信息及其估计还需要的运行时间,比如:

5.3 输出图片概览
除此之外,运行结束后,在结果文件夹fig1下,还包含了模拟结果自动分析及可视化的结果,包括速率、覆盖度、可逆度、敏感度、反应级数、主路径分析及其反应势能图和流程图等。而且单条件、一维和二维条件不同的模拟情况下其结果的展示形式也不尽相同。
5.3.1 单条件的模拟情况示例
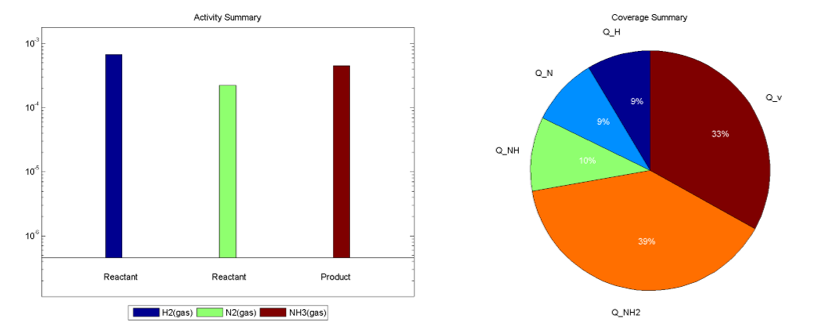
在Activity和Coverage文件夹下,分别给出了反应活性图和中间物种分布图等,如:
在Standard Free Gibbs Energy Profile和Flow Chart文件夹下,分别给出了反应势能面和反应流程图等,如:
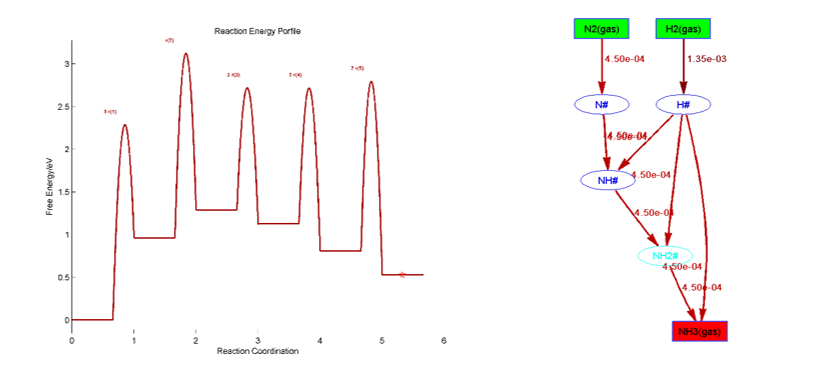
在Degree of Rate Control文件夹下,给出了主要的反应能垒、中间体的敏感度和反应级数等,如:
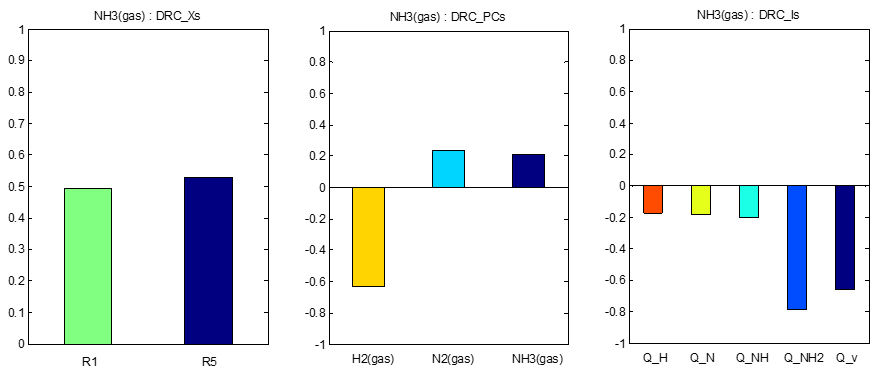
5.3.2 一维条件的模拟情况示例
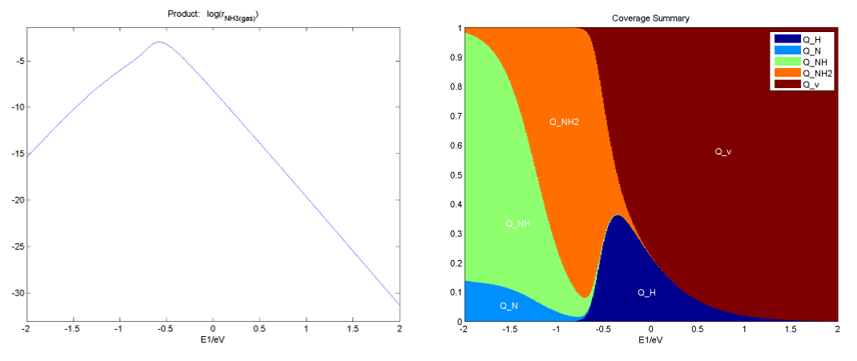
在Activity和Coverage文件夹下,分别给出了二维反应活性图和中间物种分布随一维能量描述符的变化图等,如:
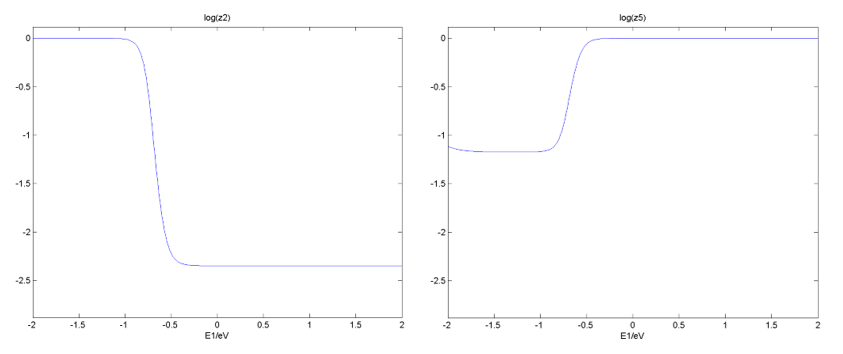
在Reversibility文件下,给出了反应步骤可逆度随一维能量描述符的变化图等,如第2和第5反应步骤:
在Standard Free Gibbs Energy Profile和Flow Chart文件夹下,分别给出了反应势能面和流程图等,在给定三个描述符能量的情况对应如下:
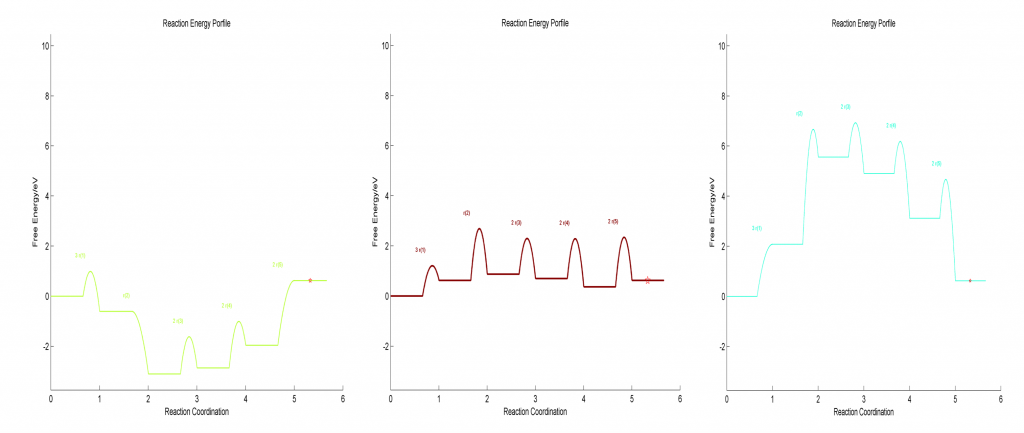
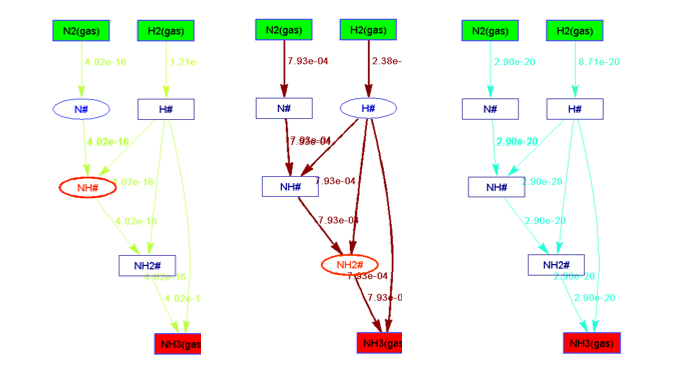
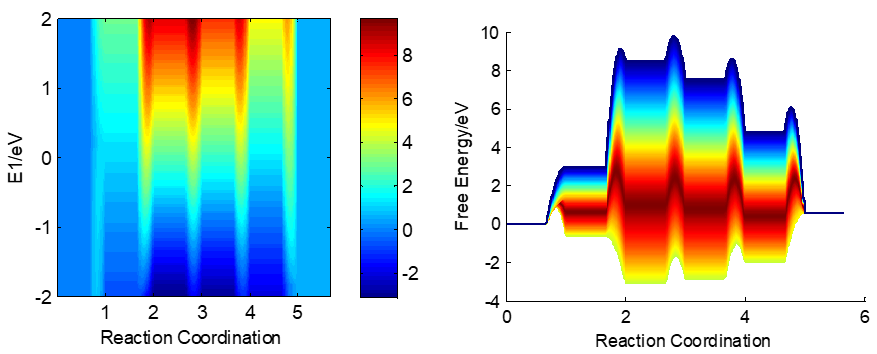
在标准压力下,整个能量描述符范围的反应势能面如下图所示:
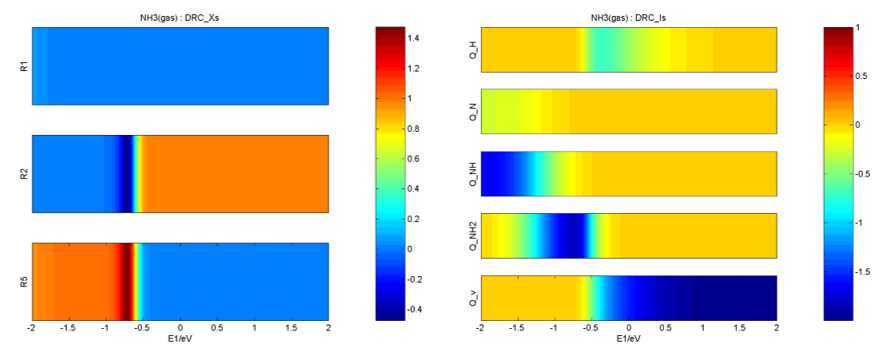
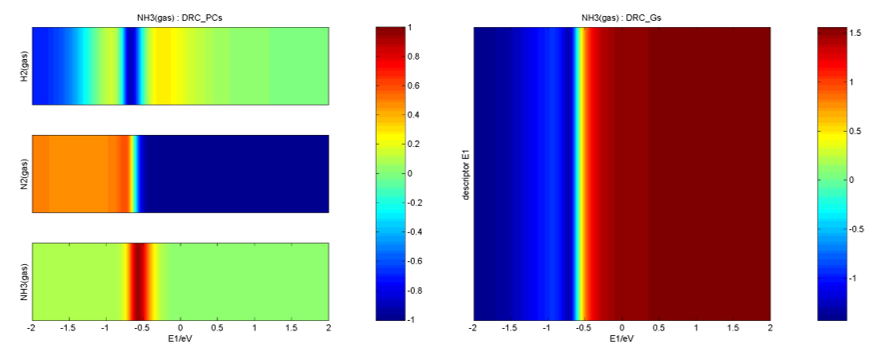
在Degree of Rate Control文件夹下,给出了主要的反应能垒、中间体的敏感度、反应级数、描述符E1的梯度随一维能量描述符变化的等高线图等,如:
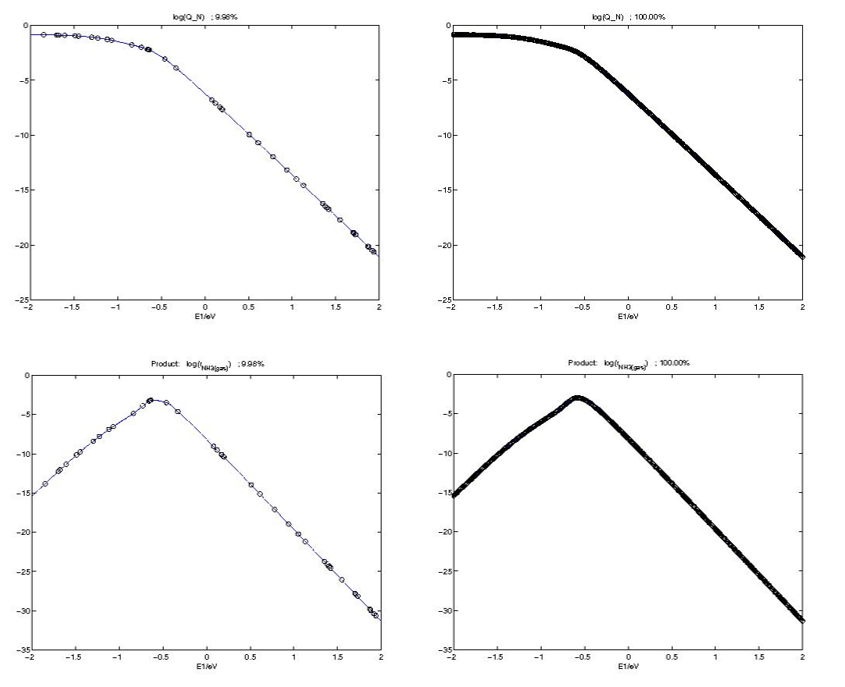
在Prediction文件夹下,包含了覆盖度和活性的不同求解比例下的二维曲线预测结果,可以看到就算只有~10%的比例,已经可以给出很好的趋势和结果。
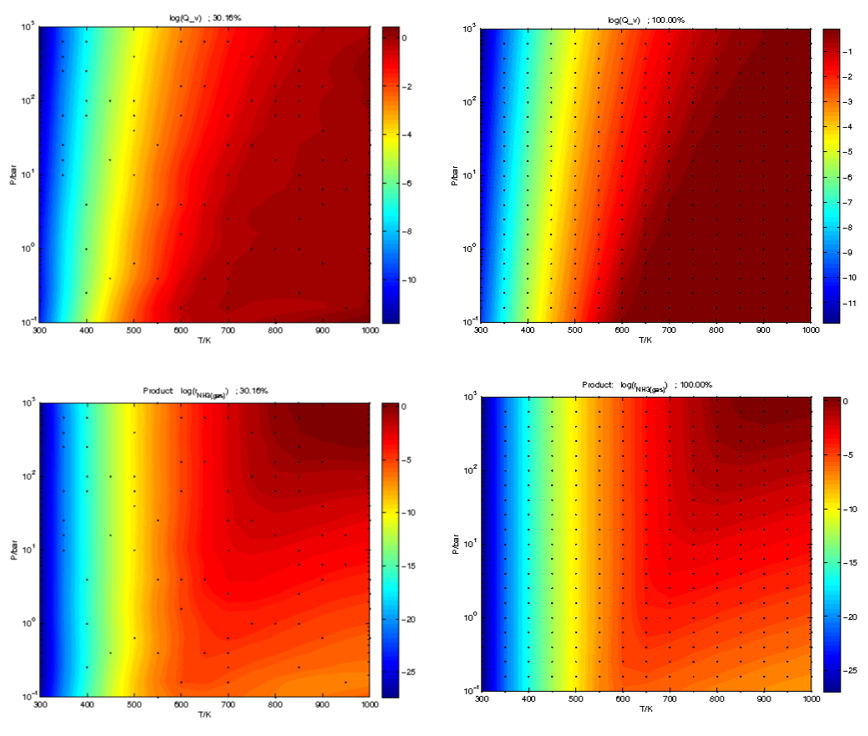
5.3.3 二维条件的模拟情况示例
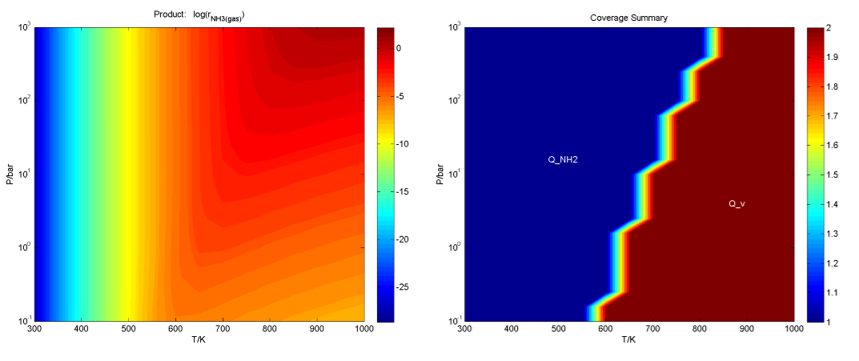
在Activity和Coverage文件夹下,分别给出了三维反应活性图和中间物种分布随温度、压力二维条件的变化图等,如:
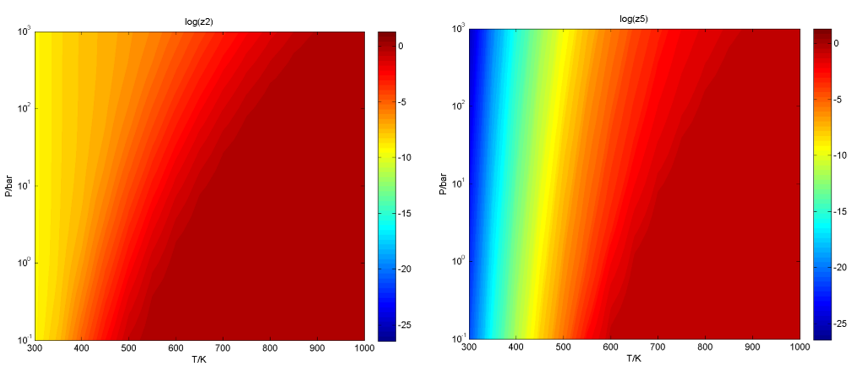
在Reversibility文件下,给出了反应步骤可逆度随温度、压力二维条件的变化图等,如:第2和第5反应步骤:
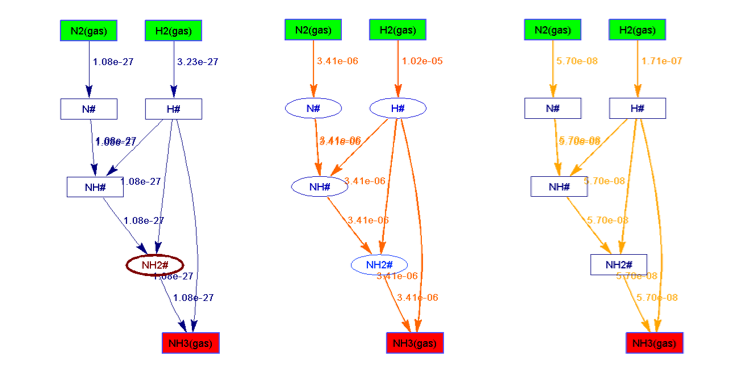
在Standard Free Gibbs Energy Profile和Flow Chart文件夹下,分别给出了反应势能面和流程图等,在给定三组温度和压力的情况对应如下:
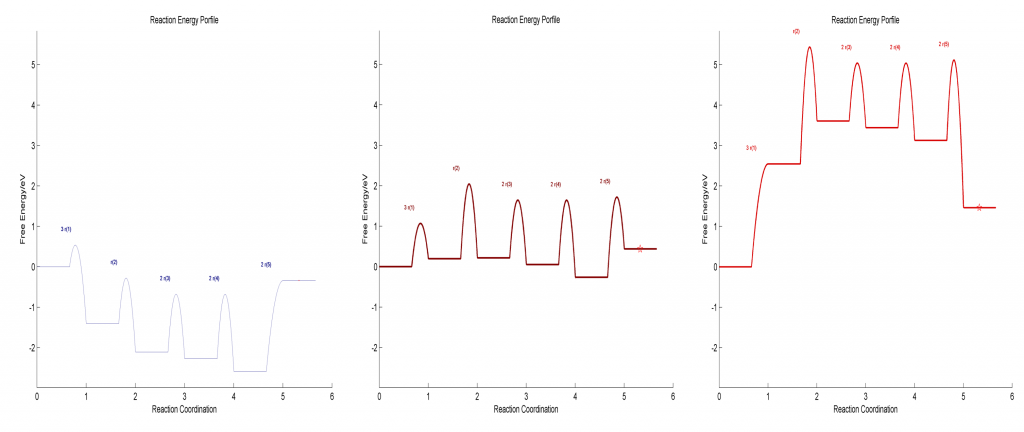
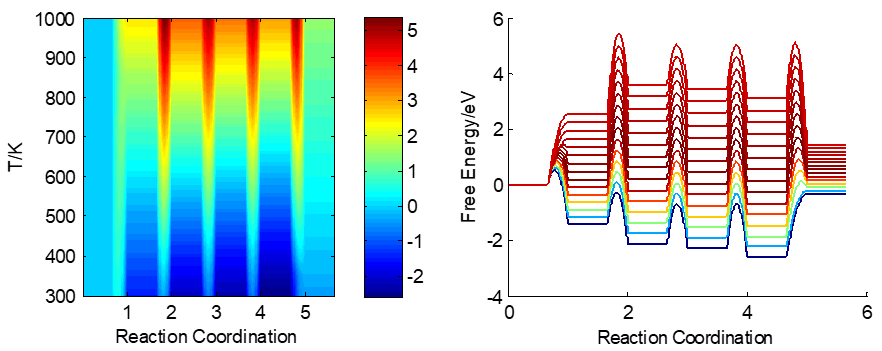
在标准压力下,整个温度范围的反应势能面如下图所示:
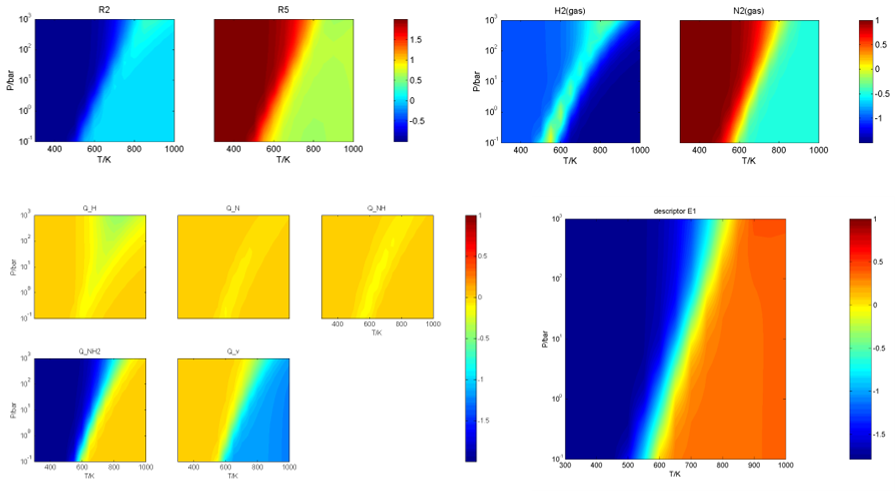
在Degree of Rate Control文件夹下,给出了主要的反应能垒、反应级数、中间体的敏感度、描述符E1的梯度随温度、压力二维条件变化的等高线图等,如:
在Prediction文件夹下,包含了覆盖度和活性的不同求解比例下的三维曲面预测结果,可以看到需要>30%的比例,就可以给出很好的趋势和结果。
4 输入文件INPUT的设置
典型的INPUT文件见第5部分模拟示例,下面我们介绍输入参数具体含义,许多参数都有可靠的默认值,当你在输入文件中遗漏掉它们时,这些默认值就变得有用(这就允许用户可以有非常短的输入文件!)。那些没有默认值选择的参数就需要用户根据具体的体系进行设置,包括反应机理、反应能量和反应条件。文件中所有的内容均为应为字符,可以采用百分号(%)或感叹号(!)作为注释标记符号,分号(;)作为分隔符,除了基元反应,所有的参数设置均采用MATLAB赋值的方式进行,即格式为:变量 = 参数值(variable = value),也允许包含一些简单的MATLAB代码、函数和命令。
4.1 反应机理
微动力学的模拟,反应的基元步骤肯定是必不可少的,它定义了微动力学的反应模型,可以包括吸附、解离、结合和脱附反应,物种包含反应物、产物、表面中间体和反应位点。
反应物种的名称需要满足变量名的命名规则,最简单的可以采用化学式书写,比如水可以用H2O表示,推荐采用标准的化学式写法,有助于反应机理的可阅读性,以及程序自动识别匹配,实现自动分子量和自带的热力学数据的计算。分别采用(gas)和(liq)来区别气相和液相物种(或分别简化为(g)和(l)或(p)和(c)),例如H2O(gas)和H2O(liq)分别表示气相和液相水,如果没有,默认物种是气相的;用井号(#)来作为反应位点,对于多个反应位点可以采用#i来表示,其中i代表第i个反应位点,例如#1和#2分别表示1号和2号反应位点;对于表面物种,采用物种名连接反应位点表示,比如吸附在1号活性位点的水为H2O#1。基元反应中,采用加号(+)分隔反应物种,采用箭头(由小于号、减号和大于号构成<->)分隔正反应逆反应,同时它也作为基元反应输入的标志。
%注释 : 基元反应 能量输入 [能垒 焓变]
(1) : H2 + 2#1 <-> 2H#1 [ 0.27 -0.20 ]
(2) : O2 + 2#1 <-> 2O#1 [ 0.85 0.47 ]
(3) : H#1 + O#2 <-> OH#2 + #1 [ 1.18 -1.09 ]
(4) : H#1 + HO#2 <-> H2O + #1 + #2 [ 1.43 -1.30 ]
以上就是氢气在金属氧化物上燃烧生产水(总反应2H2 + O2 <-> 2H2O)的4步基元反应,其中位点#1代表金属位点,位点#2代表O空位(即O#2代表表面氧位点)。基元反应中冒号(:)前面的内容将只作为注释内容,方括号中的内容为对应的能量输入,具体说明见4.2部分。
4.2 反应能量
有了基元反应步骤,需要进一步提供对应的吉布斯能垒(Ea)和反应自由能(G0),可以输入的能量也可以是反应能垒(Eb)和焓变(H0),然后设置相应的参数来自动进行处理和校正,程序内置了常见分子的热力学焓变、热容、熵(基于Shomate方程)、零点能(基于振动频率)校正的数据,对于复杂的分子,您可以自己手动校正好能量数据直接输入,或者提供相应的热力学参数输入;对于吸附/脱附的能垒,可以选择采用过渡态、碰撞理论、反应热力学等处理方式。
4.2.1 能量输入方式
1 通过设置关键字fun2GetGE= @funName提供自定义的MATLAB函数来计算对应的反应能量信息,其中funName为自定义的MATLAB函数名,可以允许自定义复杂的函数形式,包括考虑覆盖度效应等;
2 直接在基元反应的右侧提供反应能量信息,放在方括号[]中。对于单催化剂模拟,直接提供吉布斯能垒和反应自由能([Ea G0]);对于多催化剂模拟,可以提供基于能量描述符(E1/E2)的能垒和焓变的BEP或线性关系([Ea(scaling) G0(scaling)]/[Ea(BEP) G0(scaling)]),对应的需要提供(E1/E2)的模拟值;
3 直接以变量赋值的方式提供反应能量信息,比如Ea = [Ea1, …],G0 = …。
4.2.2 相关控制参数
1 E1/E2:能量描述符,最多可以允许用两个描述符(E1和E2)来模拟催化剂,设置方式采用赋值方式,比如E1 = [-1:0.1:1],E2 = linspace(-1,1,21);无默认值。
2 Fun2GetGE: 能量输入函数句柄的设置参数,fun2GetGE= @funName,用于(根据描述符)计算对应的反应能量信息,即对应函数形式为[Ea, G0] = funName(E1, E2);默认值@initGE、@initGE1和@initGE2分别对应单催化剂、1个能量和2个能量描述符的情况,需要自己提供对应的函数。
3 fun2E1/fun2E2:基于原能量值E1计算新能量值E1的自定义MATLAB函数(用于考虑覆盖效果等),比如:fun2E1 = @funName,对应函数E1 = funName(E1),或直接使用fun2E1 = @(E1) E1 + 0.2*Q1_I;无默认值。
4 BEPMode: 能垒输入的模式是比例关系还是BEP关系,即Ea =α*E1 + b或α*H0 + b 分别对应0、1;默认值1。
5 ThermoMode:反应物或产物种的热力学校正模式,包括焓变校正(dH)、熵校正(-TdS)、零点能校正(ZPE)及其各种组合,即不校正、dH、dH-TdS、dH-TdS+ZPE、-TdS、-TdS+ZPE、ZPE和dH+ZPE分别对应0、1、2、3、4、5、6和7;默认值0,即不矫正。
6 BarrierMode:吸脱附反应能垒的校正模式,包括不处理、热力学(TD)、过渡态理论(TST)和碰撞理论(CT)处理,分别对应-2、-1、0~1、2;过渡态理论处理时允许设置成0到1的值来考虑不同熵贡献的能垒值;默认值0,即不矫正。
7 ConsMode:能垒限制的模式,包括能垒需要大于0、总能(E0)或自由能(G0)及其组合,即不处理、大于0、E0、G0、(0, E0)、(0, E0)、(E0,G0)或者所有分别对应0、1、2、3、4、5、6、7;默认值0,即不处理。
8 As:吸脱附碰撞理论(CT)处理中必须设置的活性位点面积,单位是埃平方(Å2),对于多位点反应,可以设置成行向量;无默认值。
9 Mr_Species: 吸脱附碰撞理论计算时需要的物种(Species)的相对分子质量,当物种的化学式不是标准写法时,需要自己设定;因为所有元素的相对分子质量都大于1,将其值设为-2、-1和0~1可以将该物种对应的吸脱附过程改为不处理、TD、TST处理;默认值:标准化学式为对应的相对分子质量,非标准时则为-2。
10 自定义或覆盖内置的热力学参数,可采用参数Thermo_Species直接设置物种Species对应的热力学校正参数,可以包括Thermo_Species =
= [dGc];其中,dGc为物种的校正自由能;
= [G, H0, ZPE];其中G = H0 + CpT – TdS; dGc = G – H0;
= [H, H0, CpT, TdS, ZPE, Ef]; 其中Ef 为物种的生成能;
= [A, B, C, D, E, F, G, H, freqi, Ef];其中A-H为shomate参数,freqi为频率。
除内置的常见分子,其余默认值均为0;变温情况下,可增加参数T0作为对应的参考温度;此外还可以新建一个Thermodynamics.data文件,通过关键词CF、TR、AH、FQ和EF设置(分别对应化学式、温度范围、shomate参数、频率和生成能),比如H2的热力学数据设置如下:
CF = H2;
EF = [-0.272839]; FQ = [4401];
TR = [ 298, 1000]; AH = [33.07,-11.36,11.43,-2.77, -0.16, -9.98,172.71,0];
TR = [1000, 2500]; AH = [18.56, 12.26,-2.86, 0.27, 1.98, -1.15,156.29,0];
TR = [2500, 6000]; AH = [43.41, -4.29, 1.27,-0.10,-20.53,-38.52,162.08,0];
4.3 反应条件
在动力学的速率表达式中,气相、液相和表面物种(Species)的量(概率)分别以压力(P)、浓度(C)和覆盖度(Q)表示,对于压力和浓度,是无量纲化处理的,即分别除以标准压力(P0: 0.1MPa)和标准浓度(C0: 1mol/L)。物种条件的设置采用X_ Species_Type = value的格式,其中X可以是P(压力)、C(浓度)或Qi(覆盖度)(注:位点#、位点#i和多位点#i#j#k上的物种分别以Q、Qi和Qixjxk表示(多位点中以x隔开));Species气相、液相和表面物种的名称;Type为需要设置的类型,包括初始化(INIT)、固定化(FROZ)、平衡化(EQUI)和比值化(RATE)处理;value即为设定的参数值。
4.3.1 反应条件初始化(INIT)设置
设置气相、液相和表面物种Species初始压力(P)、浓度(C)和覆盖度(Qi)的初始值,比如P_CO_INIT = 1、C_H2SO4_INIT = 0.1、Q_v_INIT = 1; 默认为0。
4.3.2 反应条件固定化(FROZ)设置
固定气相、液相物种Species压力(P)、浓度(C)的值,被固定的物种,其对应的物料守恒方程dP/dt将从稳态方程中移除,设定的参数值即为稳态时被固定的物种的压力(P)或浓度(C)值。通常可以根据实验条件中反应物及转化率的值,固定所有反应物、产物的压力和浓度,来近似或简化模拟连续流动搅拌反应器(CSTR);否则需要提供实验的条件,包括初始化条件、空速、总压力/浓度等信息(见以下参数Pn、Fgt、Cn、Flt),根据其物料守恒方程来精确求解稳态方程获得。比如P_CO_FROZ = 1、C_H2SO4_FROZ = 0.1;无默认值。
4.3.3 反应步骤平衡化(EQUI)设置
假设某一反应步骤是平衡的来近似处理,即其速率为零;若实际稳态时该反应远离平衡,则该平衡处理结果很可能是无意义的,但可分别对不同反应进行平衡处理,来对活性火山型曲线/面进行分解和分析处理;因为中间体变量与稳态方程是一一对应的,因此新加的平衡处理条件需要替换原来的某稳态方程,所以需要指定替换的稳态方程所对应的中间体,即Qi_Species_EQUI = IndexOfReaction,比如Q2_OH_EQUI = 4表示第四步基元反应平衡化处理,以其速率等零r(4) = 0替换位点#2上OH表面物种所对应的物料守恒方程dQ2_OH/dt = 0;无默认值。
4.3.4 反应温度(T)设置
单位是开尔文(K),对于单一的反应温度,直接设置其值,如T = 500 K(默认值为室温条件273.15 K);如果是模拟不同反应温度下的情况,可以将其设置为行向量,如T = [300:50:1000]表示温度从300到1000 K每隔50 K布一个点。
4.3.5 连续流动搅拌器(CSTR)模拟
在含非固定化处理的物种的压力(P)或浓度(C)时,需要进一步提供实验的条件,包括空速、总压力、总浓度等信息,否则无法进行求解,包括:
Pn:气相标准化相对总压力,Pn = Ps*Ns/Ng,单位bar;
Cn:液相标准化相对总浓度,Cn = Cs*Ns/Nl,单位mol/L;
Fgt:气相标准化空速,Fgt = Vgt/Ns,单位m3/s/mol;
Flt:液相标准化空速,Flt = Vlt/Ns,单位m3/s/mol;
其中,Ns:活性位点数,单位mol;
Ps:气相相对总压力,单位bar;
Cs:液相相对总浓度,单位mol/L;
Ng:气体分子总摩尔数:单位mol;
Nl:液体分子总摩尔数;单位mol;
Vgt:气相空速,单位m3/s;
Vlt:液相空速,单位m3/s;
这里采用标准化打包的变量数据Pn、Cn、Fgt、Flt,是因为只有这些标准化的数据改变,才会对计算结果产生影响,因此并不需要全部设置Ns、Ps、Cs、Ng、Nl、Vgt和Vlt所有的参数,而且Pn和Cn只会影响体系到达稳态的(积分)时间(其本质是用来决定反应器的体积的,与下面的布点压力P和C相独立),只有Fgt和Flt变化才会影响体系稳态的解;当只有气相或液相时只需要设置对应相的参数即可,不需要全部设置;若空速设为零,则可以计算出反应达到平衡时表面的覆盖度、反应物产物的浓度等;全无默认值。
4.3.6 不同压力或浓度模拟
模拟气相或液相某一或多物种(Species)随不同压力(P)或浓度(C)下的情况,可以通过设置压力P和浓度C参数的行向量值实现,通常情况下,压力和浓度的布点是采用指数方式,例如P = 10.^[-6:1:3],即,压力P从10-6每隔一个数量级布点到103;通过设置对应物种(Species)比值化(RATE)参数,控制哪个或哪些物种参与到压力或浓度的对应改变,例如P_H2_RATE = 2, P_O2_RATE = 1,即H2和O2将随布点压力按2:1对应改变,其余未设置的物种的压力或浓度按原来的设置保持不变;若需要设置的所有比值与初始化(INIT)设置的比值一致,可以不用再单独设置,将自动采用初始化的比值;默认值是0。
4.3.7 敏感度计算(CalcDRC)
控制是否进行敏感度(DRC)的计算,包括反应能垒(B)、中间体(I)、压力/浓度(P C)和能量描述符E1/E2(G)的敏感度;其结果矩阵的变量名在datai.mat文件中分别对应DRC_B、DRC_I、DRC_PC和DRC_G;计算敏感度通常需花费要更长的求解时间,特别是基元反应步骤、中间体个数很多的情况下;默认值0;
4.4 控制参数
4.4.1 计算求解相关的参数
1 Ndigits:用于基于可逆度迭代的改进牛顿法计算的有效数字位数,位数越大,求解的稳定性和成功的概率也大,但是耗时可能也越长;默认值是1000。
2 MaxTime:每次改进的牛顿尝试的最大时间,单位秒(s);参数值越大,单次牛顿法求解成功的概率也要高,但是耗时可能更长,相同时间内可以搜索的空间也小;默认值是3。
3 MaxOdeTime:每次时间积分模拟尝试的最大时间,单位秒(s);最大时间越大,求解成功的概率也大,但是耗时可能也越长;默认值是60。
4 TimeMode:是否自动增加每代粒子群算法(PSO)中牛顿迭代的最大时间,分别对应 > 0和0,增加最大时间MaxTime有助于提高求解成功的概率;若大于0,则每代粒子群算法中牛顿迭代的最大时间按以下公式更新:MaxTime = max(MaxTime + 3,(1 + TimeMode)* MaxTime),即每次时间增加TimeMode* MaxTime且每次至少增加3 s;默认值是0.02。
4.4.2 结果分析及可视化的相关参数
1 Tspan:时间积分仿真的时间跨度参数,单位秒(s),时间跨度越大,模拟达到稳态的概率越大,但是耗时可能也越长;默认值是[0 1]。
2 PlotType:是否绘制反应曲线/图,能量分布图,流程图和时间积分模拟,不绘制、绘制曲线/面、势能面图、流程图、时间积分模拟图、所有的以及指定的,分别对应0、1、2、3、4、5 和指定行向量[1 … 4];默认值[1:3]。
3 PlotMode:多反应条件模拟时,是否绘制反应势能面图、流程图和时间积分模拟图,不绘制、第一次、全部绘制或者制定绘制分别对应0、1、inf和指定行向量;默认值是1。
4 ProfMode:以反应总能、标准自由吉布斯能、自由吉布斯能、稳定自由吉布斯能或全部形式绘制反应势能面图,分别对应0、1、2、3和4;默认值是1。
5 PlotList:指定绘制反应结果,可以包括反应速率、选择性、可逆度、覆盖度、表观活化能、以及能垒、中间体、压力/浓度、能量描述符E1/E2的敏感度:Ri、Si_j、Zi、Yi、X[s]i_j、I[s]i_j 、PC[s]i_j、G[s]i_j,其中i /j为指定的下标/活性TOF 下标或它们的组合([s]代表对应参数基于反应物或产物的结果),例如PlotList = {‘R1+1/2*2-3′,’S1+2_3+4′,’Z-2-0.3*1+5′,’Y1+2+3′,’E3′,’B-2-3*1+5 _1+2*2-3’, ‘Is5_2′,’PC1_3′,’Gs1_3’};默认值为空({})。
6 PathOrder:指定的势能面图中反应路径的顺序列表,如PathOrder = {[1 2 3],[1 4]},每个行向量代表一个反应路径,每一个数字代表基元反应的序号;默认值为空({})。
7 PathCoord:指定的势能面图中反应路径的坐标列表,如PathCoord = {[1 2 3],[1 3]},反应坐标与上面的PathOrder一一对应,若为空,则在PathOrder非空时,自动根据物料原则对齐生成坐标;默认值为空({})。
8 PathScale:指定的势能面图中反应路径的系数列表,如PathScale = {[1 2 1/2],[1 -1]};反应系数与上面的PathOrder一一对应;若为空,则在PathOrder非空时,全部系数自动设置成1;默认值为空({})。
9 FigMode:将输出图片格式保存为.fig、.png或全部,分别对应1、2或3;.fig格式可以很方便在MATLAB里面打开查看和进一步编辑处理,.png格式可以用图片软件直接打开查看;默认值是3。
10 SimMode:是否含有反应活性位点以简化反应势能面图及流程图,分别对应0和1;通常包含反应位点的流程图网络通常非常复杂,可阅读和分析性很差,特别是基元反应步骤很多时,默认值是1,即不含反应活性位点。
11 SimValue:反应速率的阈值,用于简化反应的势能面图和流程图;即小于最大反应物和产物速率*SimValue的路径将被忽略,有助于复杂反应网络的主路径提取和分析;默认值是0.05,。
12 CompMode:比较模式,在相同或各自的速率尺度范围内绘制反应势能面图和流程图,分别对应1、0;在相图范围内作图,可以方便比较不同模拟条件下的变化趋势;默认值1。
4.4.3 其他相关参数
1 Istart:是否加载之前的参数设置和稳态方程等数据(ORGi.mat)再继续运行,分别对应1、0;加载时仅更新控制参数部分;若有未完成的任务,设为不加载时将根据INPUT所有参数重新初始化、产生基元反应表达式和稳态方程等,并且如果反应机理、反应能量或反应条件改变时,请删除对应任务的输出文件,特别是doneid_i、data*.mat等,否则,前后两次运行整合了反应机理、反应能量或反应条件不一致的结果;默认值是1。
2 npar:用于平行计算的线程数,>0 时为平行计算,临时计算文件在pari下,运行结束后将自动清空并汇总,并行结果将被汇总到datai.mat和logi_parsum文件中;并产生figi/Prediction文件夹,含有根据部分获得计算的结果,利用机器学习预测完整空间的覆盖度和活性图,可以提前预判模拟是否准确及其规律;默认值是0,即1个线程串行计算。
3 runid:指定到某一次运行计算,若该任务不存在,则开始一个新的任务;存在未完成,则继续计算任务;存在且已完成,则加载数据仅更新控制参数重新进行结果分析及可视化任务;默认值是第一个未完成的任务,若无未完成任务则开始一个新的任务。
4 SaveMode:将所有数据保存为双精度(double)或高精度符号(symbolic)类型,对应0和1;保存双精度速度非常快,需要的内存和硬盘非常小;保存成高精度符号结果速度慢,需要的内存和硬盘大,特别是布点数量很多的时候;默认值是0;
5 SaveFreq:保存计算结果的频率(串行模式下),每隔多少个运行样本保存一次,参数越大,保存的频率及耗时越小,若计算速度很快、计算条件好(内存、容量大,即不会出现内存、容量不足的情况)且稳定(不会出现MATLAB程序、计算机卡死、断电等计算异常中断的情况),可以尽量考虑设大一些,特别是将数据保存为double类型时;默认值是1。
6 SkipMode:从不过滤改进牛顿方法找到的解,或者过滤包含及全是零(全是零的解指每一个位点上只有某一物种是1,其余的均为零的情况)的解,分别对应0、1和2;通常情况下,包含零的解是没有物理意义的,所以需要过滤掉,但是如果您设置的反应机理、反应能量或反应条件就太离谱、不切合实际,则该模拟并不能保证存在有物理意义的解;试图进行这样的模拟完全是浪费时间、毫无意义的,所以模拟前、求解时、甚至是求解结束,请再三检查和确认您设置的反应机理、反应能量或反应条件等参数、软件运行的输出信息等(特别是警告信息);默认值是0。
7 CheckMode:是否检查反应的热力学,不检查、第一次或者全部检查分别对应0、1和2;如果您的反应内循环的热力学不守恒(通常是同一物种,在不同基元反应中考虑共吸附方式不同导致的,当然也有可能是您的数据处理出错),则可能出现一些奇怪、不合实际的结果,比如某些反应路径的方向等,特别是他们被包含在主反应路径中时;默认值是2。
8 CalMode:计算模拟的模式,通常情况下不需要单独设置,可以根据模拟的条件自己判断;在目前的版面里,可以允许考虑布点的变量包括能量描述符E1/E2、温度T、压力P、浓度C、气相空速Fgt和液相空速Flt(分别以0、1、2、3、4、5标记),具体对应关系说明如下:
i. CalMode = 0,对应单条件模拟情况;
ii. CalMode >= 1,对应一维条件模拟情况;具体对应关系为
E1: 1.0、T: 1.1、P: 1.2、C: 1.3、Fgt: 1.4、Flt: 1.5;
iii. CalMode >= 2,对应二维条件模拟情况;具体对应关系为
(E1, E2): 2.00、(E1, T): 2.01、(E1, P): 2.02、(E1, C): 2.03、(E1, Fgt): 2.04、(E1, Flt): 2.05、(T , P): 2.12、(T , C): 2.13、(T , Fgt): 2.14、(T , Flt): 2.15、(P , C ): 2.23、(P, Fgt): 2.24、(P, Flt): 2.25、(C , Fgt): 2.34、(C , Flt): 2.35、(Fgt, Flt): 2.45。
3 输入与输出文件概述
输入/输出文件依赖于进行动力学模拟的类型、控制参数和运行的环境等。对于单一反应条件(单点)、多反应条件(一维和二维)的模拟,输出的文件和结果的可视化的展示方式不尽相同;控制参数可以控制是否计算敏感度、是否可视化输出结果以及以何种形式(.fig,.png)输出等;以可视化Matlab命令窗口运行将实时显示运行的模块及其进度,后台运行将只会在log文件里简要显示运行的模块及其进度并定时更新。
3.1 输入文件
运行程序之前,我们需要构建输入文件,默认文件名为INPUT,当然我们可以任意命名和拓展名(如.m,.in等,也可不包含拓展名),只要这个文件名的字符串满足变量名的命名规则(即变量名只能是字母a-zA-Z,数字0-9,下划线_的组合,并且之间不能包含空格,数字不能放在变量名首位,如CATKINAS.in),只需要在执行的时候作为输入变量即可,即CATKINAS CATKINAS.in或者CATKINAS(‘CATKINAS.in’);输入文件名可包含路径,若路径含有空格,请将输入文件名放在单引号中。
3.2 输出文件
开始计算后,将产生输出文件,输出文件一般在result_1文件夹下(对应默认输入文件名INPUT),如果采用其他输入文件名,输出文件夹名将为result_X格式,其中X为输入文件名不含拓展名及字符串INPUT或INPUT_开头部分,比如文件名INPUTa、INPUT_b.m、INPUT__c.CATKINAS和CATKINAS.in,其输出文件夹名将分别对应输出文件夹名result_a、result_b、result_c和result_CATKINAS。
在输出文件夹下,输出结果在logi中,其中编号i为同一输入文件第i次新的运行的结果;输出的数据保存在datai.mat中,可用于matlab快速加载、进一步分析和可视化处理;对于并行计算,将产生pari临时文件夹,运行结束将自动清空并汇总,并行结果将被汇总到logi_parsum文件;Prediction文件夹下,含有根据部分获得计算的结果,利用机器学习预测完整空间的覆盖度和活性图,可以提前预判模拟是否准确及其规律;figi文件夹中是模拟结果的可视化,包括活性、覆盖度、可逆度、敏感度、反应势能图和流程图等;任务结束后将产生logi_finished文件。
此外,执行CATKINAS –help,将显示迷你版的输入输出介绍,帮助信息内容也将保存在输入文件目录下的ReadMe文件中。
2 初始化
2.1 如何获得CATKINAS
您可以从以下网址下载:
https://www.catkinas.com/downloads
在下载页面,分别有CATKINAS.p程序、示例和手册文件。
2.2 版权应用
无论您什么时候使用CATKINAS,在所有出版物或者报告中,必须在原文中指明,例如以下方式:
The microkinetic simulation and analysis were performed using the CATKINAS code [1-3], which is developed by JianFu Chen et al.
[1] J. F. Chen, M. L. Jia, P. Hu, H. F. Wang, J Comput. Chem., 2021, 42, 379-391
[2] J. F. Chen, M. L. Jia, P. Hu, H. F. Wang, J Chem. Phys., 2021, 154(2), 024108
[3] J. F. Chen, Y. Mao, H. F. Wang, P. Hu, ACS catal., 2016, 6(10), 7078-7087
查阅输出文件log获得更多重要的引用文献。
2.3 运行CATKINAS的机型配置
CATKINAS可以运行于任何平台——所需要的只是一个能运行MATLAB R2014a及以上版本软件的CPU,包括集群和超算服务器等【需要特别注意的是,测试过CATKINAS在MATLAB R2014a、R2017a、R2019a和R2021a本机版以及R2021a/b、R2022a/b online版上均能正常运行,其他未测试过的版本可能因MATLAB自带函数更新变化可能导致CATKINAS运行出现不可预期的错误】。
2.4 如何安装CATKINAS
当你下载了CATKINAS.p的文件后,该程序可以直接在MATLAB R2014a及以上版本软件里面直接运行,不需要另外安装。
2.5 如何运行CATKINAS
为了运行CATKINAS,您必须安装MALTAB R2014a(推荐)及以上版本软件。开始您的计算时,根据您想要做的模拟,从实例中找到类似的例子,然后从编辑输入文件INPUT开始,对关键字的详细说明见第4部分。然后切换到输入文件路径下,在MATLAB命令窗口中输入:
CATKINAS INPUT
如果CATKINAS与输入文件不在同一个目录,请先将CATKINAS的路径添加到MATLAB路径变量中,可通过MATLAB可视化窗口的Set Path按钮手动添加,或者在CATKINAS路径下执行以下MATLAB代码实现:
ScriptPath = pwd;
CurrentPath = path;
path(ScriptPath, CurrentPath);
或者以后台命令运行CATKINAS,先将
ScriptPath = ‘CATKINAS-ScriptPath‘;(路径信息请对应更改)
CurrentPath = path;
path(ScriptPath, CurrentPath);
CATKINAS INPUT;
写入一个主程序文件Main.m(如果CATKINAS与输入文件在同一个目录,前三行请省略),在Linux或者Unix系统shell窗口中输入以下命令:
nohup /usr/home/MATLAB/R2014a/bin/matlab < Main.m > print-out &
在Window系统cmd窗口中输入以下命令:
“D:\Program Files\MATLAB\R2014a\bin\matlab.exe” -nodesktop -nodisplay -nosplash -r “run(‘Main’)”
执行前请对应更改matlab的安装路径信息。也可以将对应的命令内容保存到文件里面再执行,即shell的bash脚本文件或dos的bat批处理文件。
当然上面的操作都是在输入文件路径下进行的,我们也可以选择在CATKINAS程序路径下操作,只需把输入文件的完整路径和名称以参数的形式提供给CATKINAS,如:CATKINAS(‘INPUT路径/INPUT名称’);程序将根据INPUT路径自动切换并执行,输出的文件也将在该路径下。
如果在运行的过程中出现中断,包括用户取消、关闭、MATLAB卡死、内存不足、硬盘出错等问题,可以重新运行程序,程序将自动从上一次保存结果的基础上继续运行。
2.6 错误报告
目前CATKINAS包含近1万多行代码,正如其他大型的代码,CATKINAS在运行的过程中可能存在错误。如果您的模拟中发现运行异常,您可以将错误的详细内容,以及输入文件INPUT、输出文件log和其他相关文件发送给CATKINAS开发者和管理员(陈建富 博士, jfchen@ecust.edu.cn)。
1 CATKINAS概述
CATKINAS是一个用于构建、求解微动力学模拟与自动化分析的程序。允许您在短时间内进行大量基元反应步骤的复杂微观动力学模拟,可以同时包含不同反应条件下多维度的模拟、分析及其结果的可视化。CATKINAS是陈建富博士基于MATLAB语言自2014年开始逐步编写、发展和完善的,其稳态方程求解的核心是基于高精度符号计算引擎Mupad,因此速度更快。在单个输入文件的基础上,您可以对任何感兴趣的反应进行详细的微观动力学研究。除了这些模拟之外,还发展和集成了各种分析和可视化工具。例如,可以进行速率敏感度分析来确定速率控制步骤等。所有这些功能都可以在一个简单的输入文件的基础上进行控制。
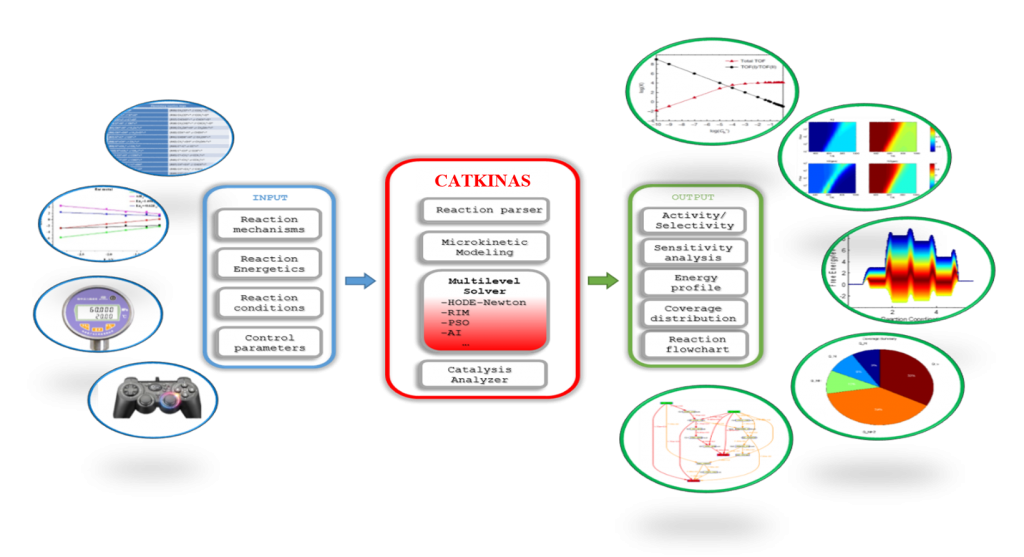
1.1 CATKINAS的特色
1 根据基元反应自动构建反应可逆度表达式、覆盖度表达式、微动力学方程和稳态方程等。
2 采用时间积分法、机器学习、粒子群优化算法等自动结合高精度的基于可逆度迭代的改进牛顿法进行求解,具有高精度、稳定性和效率。
3 包含位点守恒判断、热力学守恒校验、热力学校正、能垒处理选择和限制等功能。
4 支持单条件、多条件下活性的二维曲线及三维曲面模拟,其中条件可以包括催化剂(基于能量描述符)、反应温度、压力/浓度、反应空速等。
5 模拟结果自动分析及可视化,包括反应速率、覆盖度、可逆度、敏感度、反应级数、主路径分析及其反应势能图和流程图等。
6 根据部分计算的结果,利用机器学习预测全部计算结果,在计算的过程中可以提前洞悉活性轮廓及规律。
7 支持并行计算,支持本机、工作站、集群和超算服务器的计算。

